中等症から重症の活動性潰瘍性大腸炎患者を対象とした国内第Ⅱ/Ⅲ相試験
(1)試験概要
目的
日本人の中等症から重症の活動性※1潰瘍性大腸炎(UC)患者を対象として、ゼポジア2用量(0.46mg1日1回及び0.92mg1日1回)の有効性における用量反応性を評価し、ゼポジアのプラセボと比較した有効性を投与12週時点の臨床的改善※2によって検証する。
| ※1: | Mayoスコア6~12ポイント、内視鏡所見サブスコアが2ポイント以上、直腸出血サブスコアが1ポイント以上、かつ排便回数サブスコアが1ポイント以上の場合と定義 |
| ※2: | 完全Mayoスコアがベースラインから3ポイント以上かつ30%以上低下、かつ直腸出血サブスコアがベースラインから1ポイント以上低下又は絶対値が1ポイント以下となった場合 |
試験デザイン
多施設共同ランダム化二重盲検プラセボ対照試験

Nakase H, et al.:Gastro Hep Advances. 2026;5:100812より改変
https://creativecommons.org/licenses/by/4.0/
| a: | 生物製剤の使用歴及びステロイド使用(あり/なし)で層別化 |
| b: | 12週時点のレスポンダー(臨床的改善が認められた患者)は維持期へ移行し、12週時点のノンレスポンダー(臨床的改善が認められなかった患者)、維持期への移行後に再燃した患者、及び維持期を完了して臨床的改善を維持している患者は、オープンラベル継続投与期に移行できることとした(結果は導入期及び維持期のみを示す) |
対象
経口5-アミノサリチル酸(5-ASA)製剤又はステロイドの投与歴がある中等症から重症の活動性※1UC患者198例(プラセボ群65例、ゼポジア0.46mg群68例、ゼポジア0.92mg群65例)
| ※1: | Mayoスコア6~12ポイント、内視鏡所見サブスコアが2ポイント以上、直腸出血サブスコアが1ポイント以上、かつ排便回数サブスコアが1ポイント以上の場合と定義 |
試験方法
導入期
プラセボ群はプラセボを、ゼポジア0.46mg群では、1~4日目は0.23mg、以降は0.46mgを、ゼポジア0.92mg群では、1~4日目は0.23mg、5~7日目は0.46mg、以降は0.92mgを1日1回12週まで経口投与した。
維持期
12週時点のレスポンダーは維持期に移行し、導入期と同じ治験薬を52週まで1日1回経口投与した。
評価項目
有効性評価項目

薬力学
リンパ球絶対数、便中カルプロテクチン 等
安全性評価項目
有害事象、臨床検査値(総白血球数、好中球絶対数) 等
解析計画
有効性及びQOLの解析はITT集団(スクリーニング後にランダム化され、ゼポジアを少なくとも1回投与された全ての患者)を対象に、薬力学及び安全性の解析は安全性解析対象集団(ランダム化され、ゼポジアを少なくとも1回投与された全ての患者)を対象に実施した(データカットオフ日:2023年8月28日)。
[主要評価項目]
投与12週時点の臨床的改善率の主解析は、スクリーニング時の生物製剤の使用歴及びステロイド使用(あり/なし)で層別化したCochran-Mantel-Haenszel(CMH)検定を用いて実施した。解析結果は、臨床的改善が認められた患者の例数、割合、割合の加重差(試験結果における「プラセボ群との差」と同義)、オッズ比とその95%信頼区間及びp値で表した。
主要評価項目の主解析では、多重性を調整するため、まず両側有意水準α=0.05でゼポジア0.92mg群とプラセボ群の比較検定を実施し、統計学的に有意(p≧0.05)であった場合にのみ、0.46mg群とプラセボ群の比較検定を実施することとした。
サブグループ解析は、主要評価項目についてランダム化層別因子、患者のベースラインの背景因子及び疾患特性因子に基づき実施した。主解析と同様に生物製剤又はステロイドの使用歴により層別化したCMH検定に基づき評価され、差異と名目上のp値、両側95%信頼区間を検討した。また、スクリーニング時の生物製剤又はステロイドの使用歴に基づくサブグループ解析においては、これらの因子が除外され、患者数がITT集団の5%未満の場合、解析しないこととした。各サブグループの推定された差異をフォレストプロットで示した。事前に定義されたサブグループは以下のとおりである。

[副次評価項目]及び[探索的評価項目]
主要評価項目と同じ手法で解析した。副次及び探索的評価項目の有効性については、多重性の調整は行わず非階層的手法にて解析するため、名目上のp値とした。
[薬力学]
リンパ球絶対数:導入期及び維持期の各時点でのリンパ球絶対数(平均値及び標準誤差)を折れ線グラフとして記述した。
便中カルプロテクチン:実測値及びベースラインからの変化量については50μg/g、100μg/g、150μg/gをカットオフ値とし、分母をベースラインでカットオフ値を超えた患者数、分子を投与12週時点でカットオフ値以下であった患者数として算出し、記述的に要約した。
[安全性評価項目]
有害事象はいずれもICH国際医薬用語集(MedDRA)のVersion 26.0を用いて器官別大分類及び基本語にコード化した。
有害事象、重篤な有害事象、治験薬の投与中止に至った有害事象、及び注目すべき有害事象の発現割合を要約し、ゼポジア0.92mg群及び0.46mg群の発現割合を降順に示した。
総白血球数、好中球絶対数はベースライン時の実測値及び変化量を要約統計量(症例数、平均値、標準偏差)として示した。導入期及び維持期の来院ごとに、好中球絶対数、総白血球数に異常(好中球絶対数<1,000cells/μL、総白血球数>20,000cells/μLと定義)が認められた患者数と割合を示した。
有効性評価(改善、寛解、治癒)の定義

完全Mayoスコア:直腸出血サブスコア、排便回数サブスコア、内視鏡所見サブスコア及び医師による全般的評価サブスコアの合計
(2)患者背景
人口統計学的特性およびベースラインの疾患特性

Nakase H, et al.:Gastro Hep Advances. 2026;5:100812より改変
https://creativecommons.org/licenses/by/4.0/
5-ASA:5-アミノサリチル酸
有効性及び薬力学の結果は、承認用量である0.92mg群について示します。
(3)有効性
導入期
1) |
投与12週時点の完全Mayoスコアに基づく臨床的改善率[主要評価項目](検証的解析結果) |
投与12週時点の完全Mayoスコアに基づく臨床的改善率は、ゼポジア0.92mg群が61.5%であり、プラセボ群の32.3%よりも統計学的に有意に高く(p=0.0006、層別化CMH検定)、プラセボに対する優越性が検証されました。

2) |
投与12週時点の有効性(ITT集団)[副次評価項目] |

| 臨床的寛解率(定義2): | 直腸出血サブスコアが0ポイントかつ排便回数サブスコアが1ポイント以下(かつベースライン排便回数サブスコアから1ポイント以上低下)かつ内視鏡所見サブスコアが1ポイント以下(脆弱性なし)となった患者の割合 |
| 内視鏡的改善率: | 内視鏡所見サブスコアが1ポイント以下(脆弱性なし)となった患者の割合 |
| 粘膜治癒率: | 内視鏡所見サブスコアが1ポイント以下(脆弱性なし)かつGeboesカテゴリスコアが2.0未満となった患者の割合 |
| ※ | 生物製剤の使用歴及びステロイドの使用(あり/なし)により層別化したCMH検定に基づく |
3) |
投与12週時点の臨床的寛解率(定義1、定義3)[副次評価項目][探索的評価項目] |

| 臨床的寛解率(定義1): | 完全Mayoスコアが2ポイント以下で、かつ各サブスコアがすべて1ポイント以下となった患者の割合 |
| 臨床的寛解率(定義3): | 直腸出血サブスコアが0ポイントで、排便回数サブスコアが1ポイント以下で(かつ排便回数サブスコアがベースラインから1ポイント以上の低下を必要としない)、かつ内視鏡所見サブスコアが1ポイント以下(脆弱性なし)となった患者の割合 |
| ※ | 生物製剤の使用歴及びステロイドの使用(あり/なし)により層別化したCMH検定に基づく |
4) |
投与12週時点の組織学的寛解率[探索的評価項目] |
投与12週時点の組織学的寛解率は以下のとおりでした。

5) |
投与12週時点の臨床的改善率(導入期)[サブグループ解析] |
導入期における主要評価項目である臨床的改善率について事前に規定したサブグループ解析の結果は以下のとおりでした。

| 臨床的改善率: | 完全Mayoスコアがベースラインから3ポイント以上かつ30%以上低下、かつ直腸出血サブスコアがベースラインから1ポイント以上低下又は絶対値が1ポイント以下となった患者の割合 |
| 主要評価項目: | p値は生物製剤の使用歴及びステロイドの使用(あり/なし)により層別化したCMH検定に基づく。スクリーニング時の生物製剤又はステロイドの使用歴に基づくサブグループ解析においては、これらの因子が除外され、患者数がITT集団の5%未満の場合、解析しないこととした |
| a: | 「5-ASAの併用なし」については10例未満のため削除 |
維持期
6) |
投与52週時点の完全Mayoスコアに基づく臨床的改善率[副次評価項目] |
投与52週時点の完全Mayoスコアに基づく臨床的改善率は、ゼポジア0.92mg群が49.2%、プラセボ群が16.9%でした[p=0.0001(名目上のp値)、層別化CMH検定]。

7) |
投与52週時点の有効性(ITT集団)[副次的評価項目] |

| 臨床的寛解率(定義2): | 直腸出血サブスコアが0ポイントかつ排便回数サブスコアが1ポイント以下(かつベースライン排便回数サブスコアから1ポイント以上低下)かつ内視鏡所見サブスコアが1ポイント以下(脆弱性なし)となった患者の割合 |
| 内視鏡的改善率: | 内視鏡所見サブスコアが1ポイント以下(脆弱性なし)となった患者の割合 |
| 粘膜治癒率: | 内視鏡所見サブスコアが1ポイント以下(脆弱性なし)かつGeboesカテゴリスコアが2.0未満となった患者の割合 |
| ※ | 生物製剤の使用歴及びステロイドの使用(あり/なし)により層別化したCMH検定に基づく |
8) |
投与52週時点の臨床的寛解率(定義1、定義3)[副次評価項目][探索的評価項目] |

| 臨床的寛解率(定義1): | 完全Mayoスコアが2ポイント以下で、かつ各サブスコアがすべて1ポイント以下となった患者の割合 |
| 臨床的寛解率(定義3): | 直腸出血サブスコアが0ポイントで、排便回数サブスコアが1ポイント以下で(かつ排便回数サブスコアがベースラインから1ポイント以上の低下を必要としない)、かつ内視鏡所見サブスコアが1ポイント以下(脆弱性なし)となった患者の割合 |
| ※ | 生物製剤の使用歴及びステロイドの使用(あり/なし)により層別化したCMH検定に基づく |
9) |
投与52週時点の組織学的寛解率[探索的評価項目] |
投与52週時点の組織学的寛解率は以下のとおりでした。

(4)薬力学
1) |
導入期及び維持期の各時点でのゼポジア0.92mg群のリンパ球絶対数 |
各時点でのゼポジア0.92mg群のリンパ球絶対数は以下のとおりでした。導入期において投与開始後いずれかの時点でリンパ球絶対数が0.5×109/L未満となった患者は、プラセボ群で2例(3.1%)、ゼポジア0.92mg群で38例(58.5%)でした。

2) |
導入期の便中カルプロテクチン改善患者数 |

(5)安全性
導入期(投与0週~12週)
| ● | 投与12週までの副作用発現頻度は、ゼポジア0.92mg群で23.1%(15/65例)、ゼポジア0.46mg群で14.7%(10/68例)、プラセボ群で13.8%(9/65例)でした。主な副作用は、ゼポジア0.92mg群で肝機能異常、ALT増加、AST増加が各3.1%(2/65例)、ゼポジア0.46mg群で肝機能検査値上昇2.9%(2/68例)、プラセボ群で頭痛が3.1%(2/65例)でした。 |
| ● | 重篤な副作用は、ゼポジア0.46mg群で潰瘍性大腸炎が1例、プラセボ群で間質性肺疾患が1例に認められました。本試験の導入期においてゼポジア0.92mg群では報告されませんでした。 |
| ● | 投与中止に至った副作用は、ゼポジア0.92mg群で3例(ALT増加とAST増加が同時に発現した2例、薬物性肝障害が1例)、ゼポジア0.46mg群で潰瘍性大腸炎が1例、プラセボ群で間質性肺疾患が1例に認められました。 |
| ● | 本試験では、ゼポジア0.92mg群、ゼポジア0.46mg群及びプラセボ群において死亡に至った副作用は報告されませんでした。 |
導入期及び維持期(投与0週~52週)
| ● | 投与52週までの副作用発現頻度は、ゼポジア0.92mg群で32.3%(21/65例)、ゼポジア0.46mg群で20.6%(14/68例)、プラセボ群で13.8%(9/65例)でした。主な副作用は、ゼポジア0.92mg群でALT増加4.6%(3/65例)、γ-GTP増加、AST増加、肝機能検査値上昇、肝機能異常、帯状疱疹、回転性めまいが各3.1%(2/65例)、ゼポジア0.46mg群で、γ-GTP増加5.9%(4/68例)、肝機能検査値上昇、頭痛が各2.9%(2/68例)、プラセボ群で頭痛が3.1%(2/65例)でした。 |
| ● | 重篤な副作用は、ゼポジア0.46mg群で潰瘍性大腸炎が1例、プラセボ群で間質性肺疾患が1例に認められました。本試験の導入期及び維持期においてゼポジア0.92mg群では報告されませんでした。 |
| ● | 投与中止に至った副作用は、ゼポジア0.92mg群で4例(ALT増加とAST増加が同時に発現した2例、薬物性肝障害、黄斑浮腫が各1例)、ゼポジア0.46mg群で潰瘍性大腸炎が1例、プラセボ群で間質性肺疾患が1例に認められました。 |
| ● | 本試験では、ゼポジア0.92mg群、ゼポジア0.46mg群及びプラセボ群において死亡に至った副作用は報告されませんでした。 |
好中球絶対数及び総白血球数への影響
ゼポジア0.92mg群における維持期のベースラインからの白血球平均変化量は-2.0×109/L、プラセボ群は-0.2×109/Lでした。ベースラインからの好中球平均変化量はそれぞれ-0.9×109/L、-0.3×109/Lでした。
投与12週及び52週時点の総白血球数及び好中球絶対数のベースラインからの変化量

ゼポジア0.92mg群の導入期、維持期におけるベースライン時の好中球絶対数が1,000cells/mL未満だった患者はそれぞれ1例に認められ、本試験においてベースライン時及びベースライン後(全体)の総白血球数が20,000cells/μLを超える患者は、いずれも認められませんでした。
好中球絶対数、総白血球数の異常

副作用発現状況:導入期(安全性解析対象集団)
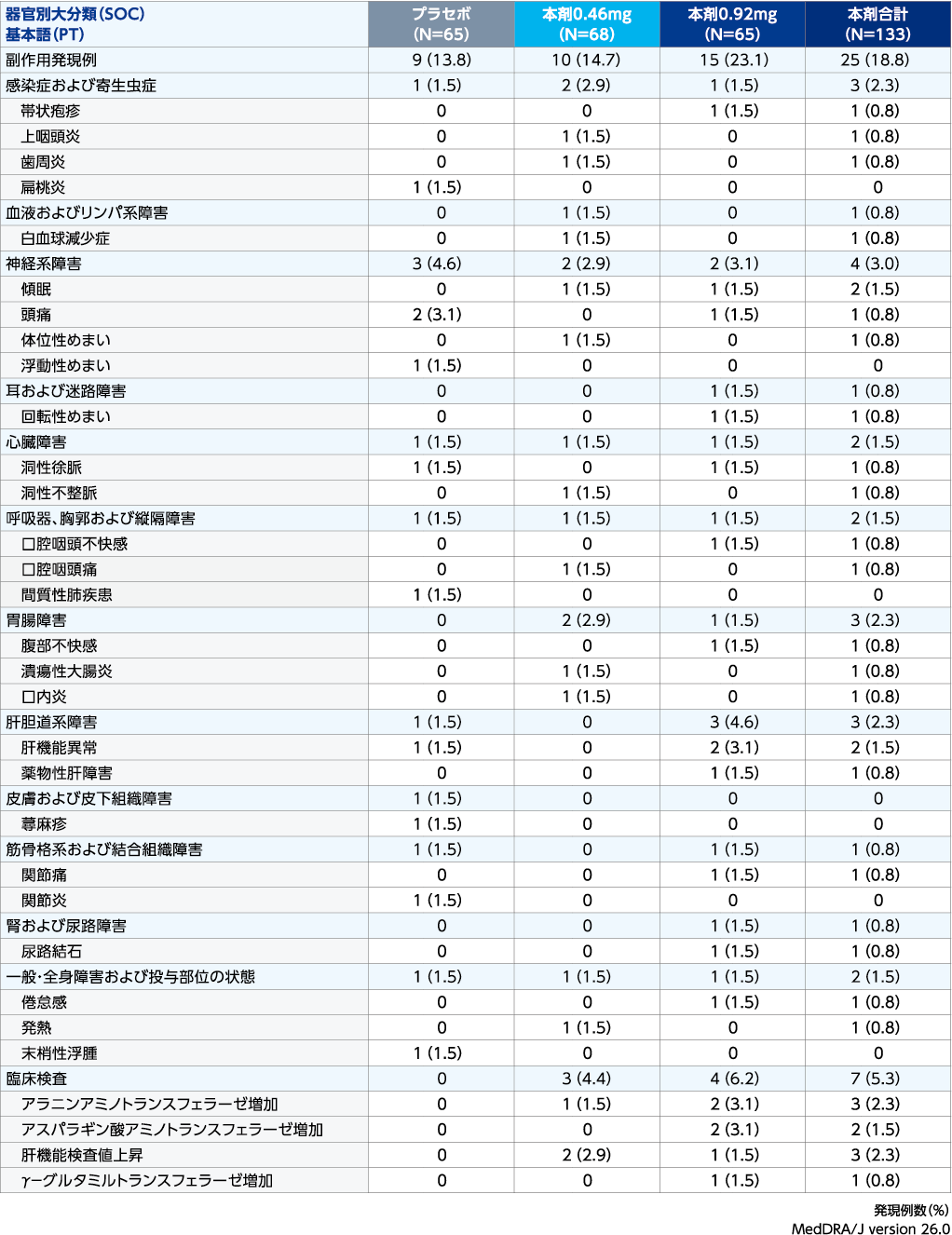
| ※ | 治験薬の初回投与日以降に発現した事象、又は初回投与日時点で継続中であり、治験薬の初回投与日から最終投与の90日後までに重症度が悪化した有害事象。1件以上の有害事象が報告された患者は1例として集計した。 同一患者で同一事象が複数回報告された場合は、当該事象の各器官別大分類/基本語内で1回のみカウントした。 |
副作用発現状況:導入期+維持期(安全性解析対象集団)

| ※ | 治験薬の初回投与日以降に発現した事象、又は初回投与日時点で継続中であり、治験薬の初回投与日から最終投与の90日後までに重症度が悪化した有害事象。1件以上の有害事象が報告された患者は1例として集計した。 同一患者で同一事象が複数回報告された場合は、当該事象の各器官別大分類/基本語内で1回のみカウントした。 |
0.46mg1日1回(0.46mg群)は用法及び用量外です。
| 6. | 用法及び用量 通常、成人にはオザニモドとして1~4日目は0.23mg、5~7日目は0.46mg、8日目以降は0.92mgを1日1回経口投与する。 |
| 8. | 重要な基本的注意(抜粋) |
| 8.4 | 本剤の薬理作用により循環血中のリンパ球数が減少するため、本剤投与開始前に血液検査(血球数算定等)を行うとともに、投与中には定期的に血液検査(血球数算定等)を行うこと。本剤投与開始後、リンパ球数が200/mm3未満となった場合には投与を中断して、患者の状態を慎重に観察し、感染症の徴候に注意すること。投与再開は、リンパ球数500/mm3以上を目安とし、治療上の有益性と危険性を慎重に評価した上で判断すること。[9.1.2、11.1.1、11.1.6参照] |